
2025-11-07 Posted by TideChem view:534
Amino acids serve as the fundamental building blocks of peptide drugs and play a critical role in the field of biomedicine. However, the inherent properties of natural amino acids limit their performance in therapeutic applications, primarily manifested in the following aspects:
(1) Poor metabolic stability: Susceptibility to proteolytic degradation, leading to a significantly shortened half-life;
(2) Limited biomembrane permeability: Particularly, charged amino acids (e.g., Arg, Lys) and polar amino acids (e.g., Asp, Glu) struggle to traverse membranes effectively, resulting in extremely low oral bioavailability. Consequently, the vast majority of peptide drugs based on natural amino acids can only be administered via injection;
(3) Limited structural diversity: Inability to support diverse peptide architectures, thereby restricting the chemical space and functional expansion of peptide drugs.
Indeed, unnatural amino acids (UAAs), which break the constraints of their natural counterparts, are reshaping the landscape of drug discovery. According to the latest 2024 research data published in the Journal of Medicinal Chemistry, FDA-approved drugs incorporating UAAs have surpassed the milestone of 100, with 44% achieving oral administration and 48% utilizing parenteral routes (including intravenous, intramuscular, and subcutaneous injections). These innovative therapeutics have been successfully applied across multiple disease areas, including infectious diseases, cancer, cardiovascular, and endocrine disorders. This trend unequivocally demonstrates that UAAs, through their unique structural tunability, are providing innovative solutions to overcome the inherent limitations of peptide drugs and driving the development of a new generation of biotherapeutics.
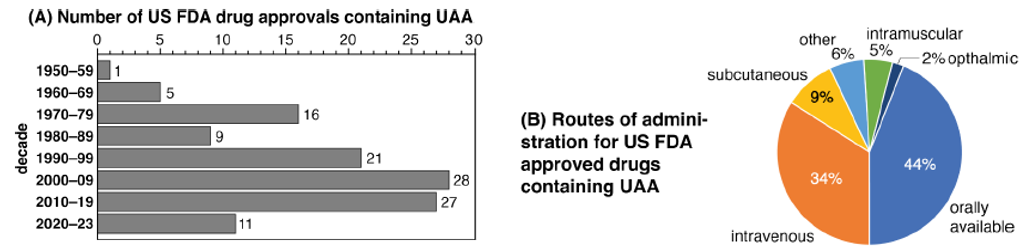
Figure 1. (a) Decade-wise distribution of UAAs containing US FDA approved drugs; (b) Comparison between various routes of administration.
Peptide drugs are susceptible to recognition and degradation by widely distributed proteases and peptidases in vivo. Their structure, primarily composed of natural L-amino acids linked by peptide bonds, presents an ideal substrate for various proteolytic enzymes, leading to rapid inactivation in the bloodstream and tissues. Furthermore, the molecular weight of most peptides falls well below the renal filtration threshold (approximately 30 kDa), facilitating their rapid clearance from circulation via glomerular filtration. Additionally, certain peptides may elicit immune responses, resulting in neutralization by antibodies and subsequent loss of activity. These combined mechanisms underlie the characteristically short in vivo half-life of peptide drugs.
Gonadotropin-releasing hormone (GnRH) is a decapeptide hormone secreted by the hypothalamus that regulates reproductive functions. Its primary physiological role is to stimulate the synthesis and secretion of gonadotropins in the pituitary gland. The His2 and Trp3 residues constitute its bioactive core, while the Arg8 is critical for GnRH's ability to regulate gonadotropin synthesis and release. The peptide bond between Gly6 and residues at Tyr5 and Leu7 is highly susceptible to cleavage by endopeptidases. Similarly, the linkage between Pro9 and Gly10 is readily cleaved by carboxyamidases. Due to this rapid enzymatic degradation, GnRH has an extremely short half-life of only 2-4 minutes in the systemic circulation.
Leuprolide, a GnRH agonist developed by AbbVie, received FDA approval in 1985 for treating conditions such as central precocious puberty and prostate cancer. Its structural modifications—replacing Gly6 with D-Leu6 and the C-terminal glycinamide with ethylamide—significantly enhanced the drug's enzymatic stability. These changes contributed to an extended plasma half-life of 3-4 hours.
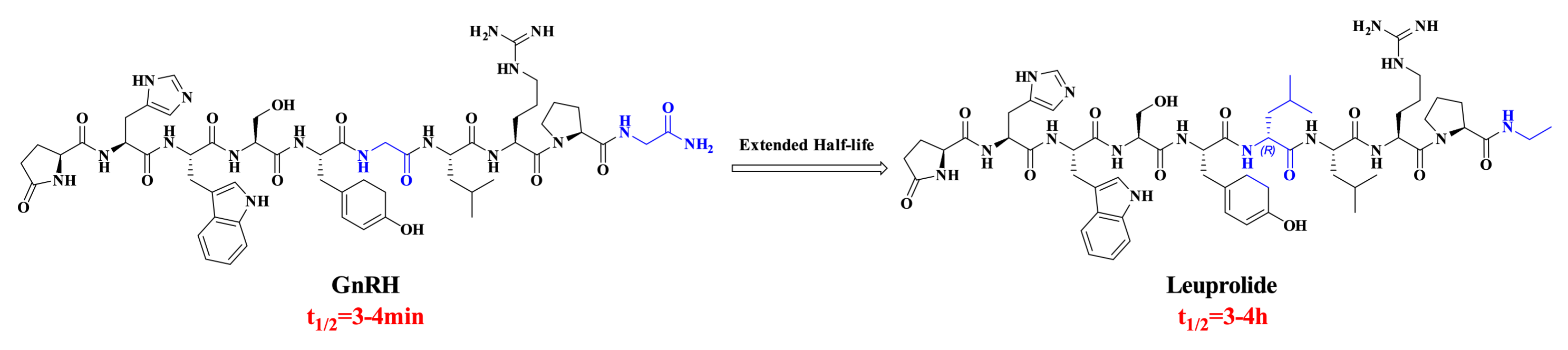
Figure 2. UAAs peptides with Extended Half-life.
The incorporation of unnatural amino acids serves as a key strategy for optimizing peptide therapeutics by enhancing their target affinity and selectivity. Specifically, introducing conformationally restricted unnatural amino acids enables the locking of the peptide into its optimal binding conformation for the target receptor, thereby significantly improving binding affinity.
Neurotensin (NT) mediates analgesia by activating the G protein-coupled receptors NTS1 and NTS2. However, activation of NTS1 is associated with adverse effects such as hypotension and hypothermia. Therefore, enhancing drug selectivity for NTS2 over NTS1 is a key strategy for developing potent analgesic candidates with fewer side effects.
Structural modifications were performed on the bioactive natural neurotensin fragment NT(8-13) (Arg8-Arg9-Pro10-Tyr11-Ile12-Leu13)): Arg9 was substituted with Lys9, while Arg8, Tyr11, and Ile12 were replaced with the unnatural amino acids β3hLys8, 6-OH-Tic11, and Tle12, respectively, yielding Compound 1 (β3hLys8-Lys9-Pro10-(6-OH)Tic11-Tle12-Leu13). This compound is a potent and NTS2-selective analogue, exhibiting an approximately 1,000-fold improvement in NTS1/NTS2 selectivity compared to the native fragment. In a rat formalin-induced pain model, intrathecal administration of the analogue demonstrated significant analgesic activity, with an ED₅₀ of 1.4 nmol. Furthermore, intravenous administration did not induce persistent hypotension or hypothermia.
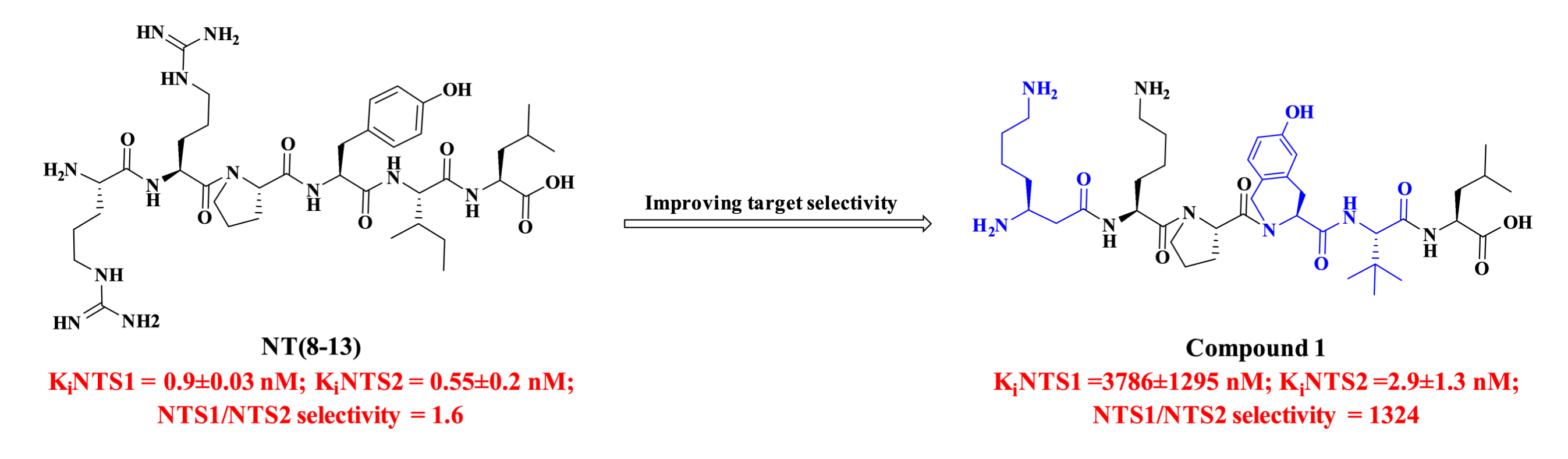
Figure 3. UAAs peptides with improved target selectivity.
The majority of peptide drugs low oral bioavailability, often necessitating injection-based administration, which leads to poor patient compliance and significantly hinders their clinical development. In vivo proteases and peptidases readily recognize and degrade peptides with extended and flexible conformations. Strategies such as incorporating D-amino acids, N-methylated amino acids, or constructing cyclic structures can enhance the rigidity of the peptide backbone and restrict conformational flexibility, thereby effectively resisting enzymatic degradation and significantly improving oral bioavailability.
N-Methylation of peptide compounds reduces their capacity to form hydrogen bonds, thereby contributing to improved oral bioavailability. Following N-methylation of D-Leu2, Leu3, and Tyr6 in the cyclic peptide cyclo(L-Leu1–D-Leu2–L-Leu3–L-Leu4–D-Pro5–L-Tyr6) (Compound 1), the resulting Compound 2 exhibited significantly enhanced permeability in the parallel artificial membrane permeability assay (PAMPA), comparable to that of the oral drug propranolol. Furthermore, in a rat pharmacokinetic study, it demonstrated an oral bioavailability of 28%, similar to that of cyclosporine A.
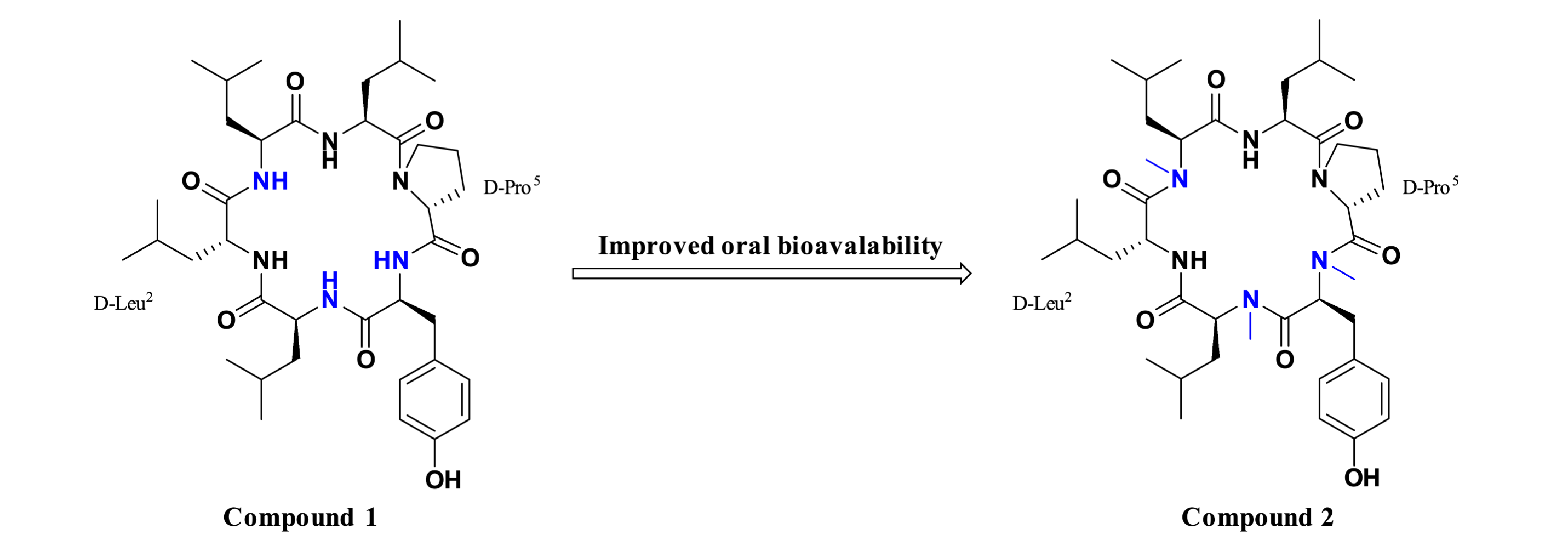
Figure 4. N-Methylated peptides with improved oral bioavailability.
The incorporation of unnatural amino acids serves as a core technology in modern peptide drug development. Through precise chemical modification, it enables systematic optimization of key peptide properties—including metabolic stability, membrane permeability, and target activity—effectively overcoming the clinical translation barriers inherent to natural peptides.
Leveraging a diverse library of over 100 structurally varied unnatural amino acids, Tide Chem offers cost-effective products and customized process solutions to fully support the R&D and production needs of innovative peptide therapeutics.
Sharma, Krishna K et al. “Unnatural Amino Acids: Strategies, Designs, and Applications in Medicinal Chemistry and Drug Discovery.” Journal of medicinal chemistry vol. 67,22 (2024): 19932-19965. doi:10.1021/acs.jmedchem.4c00110.
Räder, Andreas F B et al. “Improving oral bioavailability of cyclic peptides by N-methylation.” Bioorganic & medicinal chemistry vol. 26,10 (2018): 2766-2773. doi:10.1016/j.bmc.2017.08.031.
White, Tina R et al. “On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds.” Nature chemical biology vol. 7,11 810-7. 25 Sep. 2011, doi:10.1038/nchembio.664.
Eiselt, Emilie et al. “Neurotensin Analogues Containing Cyclic Surrogates of Tyrosine at Position 11 Improve NTS2 Selectivity Leading to Analgesia without Hypotension and Hypothermia.” ACS chemical neuroscience vol. 10,11 (2019): 4535-4544. doi:10.1021/acschemneuro.9b00390.